
Overview
The urea cycle, also known as the ornithine cycle, is a metabolic pathway in the liver that facilitates the removal of excess nitrogen from the body. Ammonia (free ammonia as ammonium ion is toxic), which is produced during the breakdown of amino acids, is converted into urea, a relatively less toxic substance that can be eliminated from the body via urination. The urea cycle comprises a series of enzymatic reactions and is subject to regulation by a variety of factors, such as substrates and the enzymatic activity of key enzymes.
Functions of the urea cycle:
1. Removal of excess nitrogen: The urea cycle is the primary mechanism for removing excess nitrogen from the body. Ammonia is toxic to cells and can cause damage to the brain and other organs if it accumulates.
2. Regulation of amino acid metabolism: The urea cycle is closely linked to the metabolism of amino acids. The cycle helps regulate the levels of amino acids in the body by controlling the amount of nitrogen that is released during their breakdown.
3. pH regulation: The urea cycle plays a role in regulating the pH of the body by producing ammonium ions, which can act as a buffer to maintain the pH of the blood within a narrow range.
4. Energy production: The urea cycle requires energy in the form of ATP to convert ammonia into urea. The cycle also produces fumarate, which can be used in the citric acid cycle to produce more ATP.
Salient feature of Urea cycle:
The urea cycle initiates in the mitochondria of hepatocytes and concludes in the cytoplasm. Urea is composed of two nitrogen atoms, one of which originates from NH4+ and the other from aspartate. The ammonium ion (NH4+) is generated through the process of deamination of amino acids. Conversely, the nitrogen found in aspartate is primarily derived from transamination reactions.
1.Ammonia is produced in the liver by the breakdown of amino acids from dietary protein or from the turnover of cellular proteins.
2. Ammonia is combined with carbon dioxide to form carbamoyl phosphate, which is catalyzed by the enzyme Carbamoyl Phosphate Synthetase I (CPSI).
3.Ornithine combines with carbamoyl phosphate to form citrulline, which is catalyzed by the enzyme Ornithine TransCarbamylase (OTC).
4.Citrulline is transported out of the mitochondria and into the cytoplasm of the liver cell.
5.In the cytoplasm, citrulline combines with aspartate to form argininosuccinate, which is catalyzed by the enzyme argininosuccinate synthetase.
6.Argininosuccinate is then cleaved to form arginine and fumarate, which is catalyzed by the enzyme argininosuccinate lyase.
7. Finally, arginine is hydrolyzed by the enzyme arginase to form urea and ornithine, which is then transported back into the mitochondria to participate in another round of the cycle (Figure:1).
| Note: Urea, which contains 2 nitrogens, is synthesized in the liver from aspartate and carbamoyl phosphate, which in turn is produced from ammonium ion and carbon dioxide by mitochondrial carbamoyl phosphate synthetase. This enzyme requires N-acetyl glutamate as an activator. N-acetyl glutamate is produced only when free amino acids are present. |
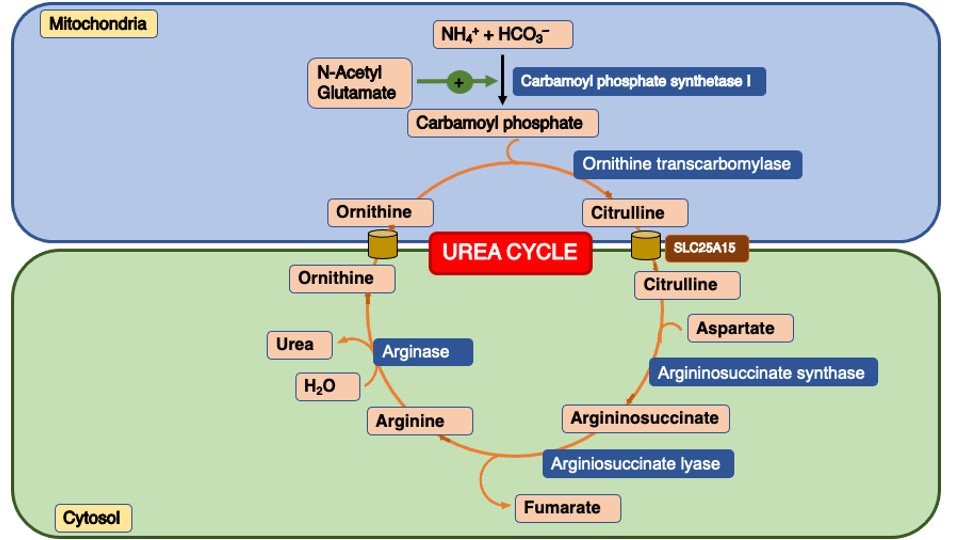
Figure 1.: Different steps of Urea Cycle
Synthesis of Urea: Occurs in hepatocytes (Mitochondria and cytosol)
The reactions are as follows:
Step 1. CO2 from bicarbonate and NH4 from the two sources mentioned above combine together in the liver mitochondria to form carbamoyl phosphate in presence of ATP and Mg2+ by the enzyme Carbamoyl phosphate synthetase I (CPSI)-Regulatory Enzyme.
Step 2. Carbamoyl phosphate reacts with ornithine transferring the carbamoyl moiety to produce citrulline: by the enzyme ornithine transcarbamylase.
Step 3. Aspartic acid and citrulline react to generate argininosuccinic acid. Argininosuccinic acid synthase is the enzyme needed.
Step 4. The enzyme Argininosuccinate lyase catalyzes the cleavage of Argininosuccinic acid into Arginine and fumarate. Fumarate is incorporated into the tricarboxylic acid cycle and subsequently utilized for ATP generation.
Step 5. Arginine is converted into urea and ornithine by the cytosolic enzyme arginase. Therefore, ornithine undergoes regeneration and can be transported into the mitochondrion to initiate another round of the urea cycle.
Energetics of the urea cycle:
The synthesis of one molecule of urea require four high energy phosphate groups (equivalent to 4 ATPs):
1. 2 ATPs used up to make up carbamoyl Phosphate
2. 1 ATP and two High-energy bonds (converted into AMP, equivalent to 2 ATP utilize and form 2 ADP) to make up Arginosuccinate.
Any reaction that creates a new C-N bond costs one ATP. However, the urea cycle also causes a net conversion of oxaloacetate to fumarate via Aspartate and regeneration of oxaloacetate produces NADH in the malate dehydrogenase reaction. Each NADH molecule can generate up to 3 ATPs during mitochondrial respiration.
Regulation of the urea cycle:
A. Short Term Regulation:
The activity of the urea cycle is regulated short term via the formation of N-acetyl-glutamate. The first enzyme carbamoyl-phosphate synthetase I (CPS I) is allosterically regulated by N-acetyl-glutamate which is synthesized from acetyl-CoA and glutamate by N-acetyl-glutamate synthase (Figure 2).
B. Long term Regulation:
Long-term regulation of liver synthesis of the four urea-cycle enzymes and carbamoyl phosphate synthetase I meet oscillations in urea cycle activity demand.
Starving and high-protein diet individuals synthesize all five enzymes more rapidly than well-fed carbohydrates and fat. Protein-free diets reduce urea cycle enzymes (Figure 2).
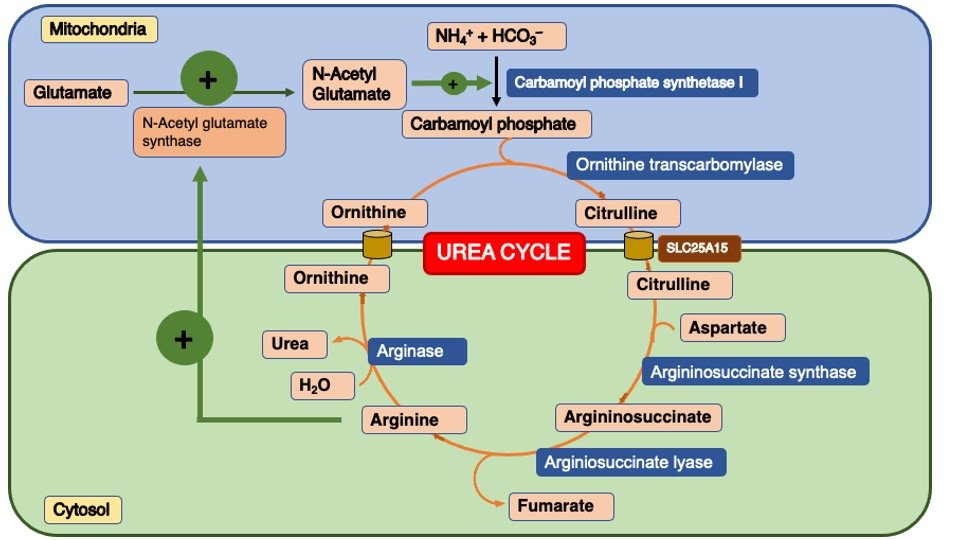
Figure 2 : Regulation of the Urea cycle
Factors influence the Urea cycle:
| S.No. | Regulation Mechanism | Description |
| 1. | Substrate availability | The rate of the urea cycle is influenced by the concentration of substrates, including ammonia, ornithine, citrulline, and arginine. The process is activated by increased concentrations of ammonia, whereas reduced levels of ammonia may impede the cycle. |
| 2. | Allosteric regulation | Several intermediates of the urea cycle exhibit allosteric regulation. It has been observed that ornithine and citrulline exert a positive regulatory effect on carbamoyl phosphate synthetase I, thereby enhancing its enzymatic activity. By inhibiting N-acetyl glutamate synthetase with arginine, carbamoyl phosphate synthetase I is indirectly regulated. |
| 3. | Hormonal regulation | The synthesis of N-acetyl glutamate (NAG), which acts as a cofactor for carbamoyl phosphate synthetase I, is triggered by an increase in arginine levels. The production of N-acetyl-glutamate (NAG) is triggered by the presence of arginine, which in turn promotes the activity of the urea cycle. Urea cycle enzymes’ expression and activity can be modulated by hormones, including glucagon and glucocorticoids. |
| 4. | Genetic regulation | The expression of genes encoding urea cycle enzymes is regulated at the transcriptional level. Transcription factors and genetic mutations can influence the production and activity of these enzymes. |
| 5. | Nitrogen balance | The urea cycle is governed by nitrogen equilibrium in the body. To remove excess nitrogen from high-protein diets, the urea cycle undergoes upregulation. In order to conserve nitrogen, fasting or a low protein intake downregulates the urea cycle. |
Clinical Note:
Genetic Deficiencies of the Urea Cycle
A combination of hyperammonemia, elevated blood glutamine levels, and decreased blood urea nitrogen (BUN) indicates a potential defect in the urea cycle. In cases of neonatal onset, infants generally appear normal during the first 24 hours. However, symptoms such as lethargy, vomiting, and hyperventilation typically emerge between 24 to 72 hours after birth. If left untreated, these symptoms can escalate to coma, respiratory failure, and ultimately, death. There are two deficiencies related to mitochondrial enzymes in the urea cycle: carbamoyl phosphate synthetase and ornithine transcarbamoylase. These can be differentiated by the increase of orotic acid and uracil, which is present in ornithine transcarbamoylase deficiency but absent in carbamoyl phosphate synthetase deficiency. Orotic acid and uracil are intermediates in the synthesis of pyrimidines. This pathway is activated by the buildup of carbamoyl phosphate, which serves as the substrate for ornithine transcarbamoylase in the urea cycle and for aspartate transcarbamoylase in pyrimidine synthesis.
Blood urea nitrogen (BUN) is an abbreviation. The physiological concentration of urea in the human body is subject to variability based on a multitude of factors, including but not limited to age, dietary habits, and renal performance. Typically, the standard range for blood urea nitrogen (BUN), or urea concentration in the blood, falls between 7 and 20 milligrammes per deciliter (mg/dL), or 2.5 to 7.1 millimoles per litre (mmol/L).
Abnormal levels of urea can indicate different medical conditions or physiological changes. Some examples include:
- Elevated Urea Levels (Hyperuremia):
- Impaired kidney function or kidney disease
- Dehydration
- High protein intake
- Gastrointestinal bleeding
- Certain medications (e.g., corticosteroids)
- Decreased Urea Levels (Hypouremia):
- Severe liver disease or failure
- Malnutrition
- Overhydration or excessive fluid intake
- Impaired liver function or low protein intake
- Uraemia is characterized by an elevated BUN level, which indicates an elevated blood urea concentration. Notably, hyperuricemia refers to an elevated concentration of urate, which is distinct from urea as a molecule.
- Azotaemia is a medical condition characterized by elevated levels of nitrogen concentration in the bloodstream, typically indicated by an increase in blood urea nitrogen (BUN) values.
- Antibodies to carbamoyl-phosphate synthetase I and arginase, immunohistochemistry distinguishes hepatocellular carcinomas from other adenocarcinomas.
- Urea can be reabsorbed by the kidney tubules.
- The BUN/creatinine ratio, measured in mg/dL, typically ranges from 10 to 20 in individuals who are considered to be in good health. Reduced urea absorption due to renal impairment lowers the ratio below 10. However, a ratio greater than 20 indicates dehydration, which decreases filtration.
- The capacity of individuals with advanced liver disease to produce urea may be reduced, leading to a decrease in the level of BUN. Conversely, an individual who is undergoing renal failure may demonstrate a reduced capacity to excrete urea, which can lead to increased levels of BUN and the emergence of uremia or uremic syndrome.
An acute episode characterized by progressive fatigue, emesis, convulsions, and unconsciousness typically indicates inadequate nitrogen excretion via the urea cycle. The first sign of hyperammonemia is respiratory alkalosis. Ornithine, citrulline, argininosuccinate, arginine, homocitrulline, and orotate are typically measured in the blood and urine to diagnose urea cycle abnormalities. Homocitrulline resembles citrulline, but has an extended aliphatic chain with an additional–CH2–group. Condensation of carbamoyl phosphate and lysine can produce homocitrulline. Orotate is produced when excess carbamoyl phosphate enters the cytosol from the mitochondria.
Ammonia Toxicity (encephalopathy):
Ammonia is a universal participant in amino acid synthesis and degradation. But its accumulation > 25–100μg/dl becomes toxic mainly to the central nervous system (CNS). The reasons for toxicity of ammonia to CNS are as follows
1) The major toxic effects of ammonia in the brain probably involve changes in cellular PH and depletion of certain TCA cycle intermediates.
2) More and more ammonia might deplete α-ketoglutarate, an intermediate in TCA cycle to form Glutamate.
3) More and more glutamate might undergo decarboxylation to form Gamma-amino butyric acid (GABA), an inhibitory neurotransmitter that inhibits ATP synthesis and accounts for the slurred speech and bizarre behaviours.
4) If the concentration of ammonia builds up it creates osmotic pressure by combining with H2O leading to coma.
Inherited and acquired Urea-Cycle defects:
Ammonia intoxication can be caused by inherited or acquired defects in ammonia trapping or in urea cycle; most of the inhabited defects occur at a rate of 1 in every 30,000 births. Inherited defects in the urea–cycle enzyme result in mental retardation.
Table: Inherited defects in the urea cycle:
| Disease Hyperammonemia | Defective Enzyme | Produce excessive amounts of |
| Type I | CPS I | Ammonia |
| Type II | Ornithine transcarbamylase | Ammonia |
| Citrullinemia | Argininosuccinate Synthase | Citrulline |
| Argininosuccinic | Argininosuccinate Lyase | Argininosuccinic aciduria |
| Argenemia | Arginase | Arginine |
Note: Ammonia intoxication caused by inherited defects in the urea cycle enzyme after argininosuccinate synthase can be treated by a diet low in protein and amino acid and supplemented by Arginine and citrulline.
Treatment with sodium benzoate can produce additional disposal of non-urea nitrogen by combining with glycine the product hippuric acid, is excreted in the urine. Sodium phenyl lactate is even more effective, since it condenses with glutamine, the major carrier of excess Nitrogen. The resulting Compound phenylacetylglutamine is excreted carrying two nitrogen’s with it.
Another mechanism for the treatment of defects in the urea cycle is the administration of ketoacids.
Acquired urea cycle abnormalities:
Any disease or condition that adversely affects liver mitochondria can also produce an increased level of ammonia in the blood such conditions include liver cirrhosis, alcoholism, hepatitis, and Reye’s syndromes.
Severe liver injury causes hyperammonemia and hepatic encephalopathy. Hepatic encephalopathy is a common complication of portal hypertension. Portal hypertension is caused by cirrhosis, which is typically caused by hepatitis B or C, alcoholic liver disease, or metabolic syndrome. Cerebrospinal glutamine and respiratory alkalosis are caused by hyperammonemia. These people’s blood may also include brain-toxic substances that the liver normally eliminates.
Chronic hepatic encephalopathy can be treated with lactulose and lactitol, sugars and sugar alcohols that only gut bacteria can break down. The laxatives lactulose and lactitol increase nitrogen excretion in the faeces while decreasing intestinal ammonia release. If lactulose or lactitol are not tolerated, rifaximin can diminish colon NH4+ producing bacteria.
| Note: The kidney contains glutaminase, allowing it to deaminate glutamine arriving in the blood and to eliminate the amino group as ammonium ion in urine. The reaction is irreversible. Kidney glutaminase is induced by chronic acidosis, in which excretion of ammonium may become the major defense mechanism. The liver has only small quantities of glutaminase; however, levels of the enzyme are high in the intestine where the ammonium ion from deamination can be sent directly to the liver via the portal blood and used for urea synthesis. The intestinal bacteria and glutamine from dietary protein contribute to the intestinal ammonia entering the portal blood. |
Measurement of Urea in laboratory:
Various methods can be employed in the laboratory to determine the concentration of urea in diverse samples. The laboratory testing commonly employs various types of urea measurements.
In the laboratory, urea can be measured using different methods to assess its concentration in various samples. Some common types of urea measurements used in laboratory testing:
- Blood Urea Nitrogen (BUN): The measurement of urea in the blood through the BUN method is commonly employed. The test measures the concentration of urea nitrogen in the bloodstream. Blood urea nitrogen (BUN) levels are frequently employed as a biomarker for assessing renal function and hydration status.
- Urine Urea: The measurement of urine urea levels is a useful tool for assessing renal function, urine concentration, and urea excretion. This aids in evaluating the renal excretory function of the body with regards to urea elimination.
- Urea Clearance: Urea clearance measures urea levels in blood and urine to estimate glomerular filtration rate, reflecting kidney function and their ability to eliminate urea from the bloodstream.
- Cerebrospinal Fluid (CSF) Urea: The measurement of urea in cerebrospinal fluid (CSF) helps evaluate urea concentration, aiding in the identification of neurological disorders and assessing its effects on the central nervous system.
- Ammonia Levels: The measurement of ammonia levels is crucial for assessing liver function and urea cycle disorders, as it results from urea metabolism. High ammonia concentrations in the blood can indicate liver dysfunction or impaired urea metabolism.
Reference book
- Harper’s Illustrated Biochemistry, Thirty-Second Edition
- Textbook of Biochemistry with Clinical Correlations Hardcover – Illustrated, 22 January 2010 by Thomas M. Devlin
- Lehninger Principles of Biochemistry: 6th Edition
- Lippincott’s Illustrated Reviews – Biochemistry South
Multiple Choice Questions
1. Which of the following is not a product of the urea cycle?
a) Urea
b) Ammonia
c) Citrulline
d) Ornithine
Answer: b
2. Where does the urea cycle occur in the body?
a) Liver
b) Kidneys
c) Lungs
d) Pancreas
Answer: a
3. Which enzyme converts ammonia and carbon dioxide to carbamoyl phosphate?
a) Ornithine transcarbamylase
b) Argininosuccinate synthase
c) Carbamoyl phosphate synthetase I
d) Arginase
Answer: c
4. Which molecule is used to transfer an amino group to carbamoyl phosphate in the first step of the urea cycle?
a) Glutamate
b) Alanine
c) Aspartate
d) Glycine
Answer: a
5. What is the function of argininosuccinate lyase in the urea cycle?
a) It converts argininosuccinate to arginine
b) It converts arginine to urea
c) It converts citrulline to argininosuccinate
d) It converts ornithine to citrulline
Answer: a
6. Which enzyme deficiency is the most common cause of urea cycle disorders?
a) Ornithine transcarbamylase deficiency
b) Argininosuccinate synthase deficiency
c) Argininosuccinate lyase deficiency
d) Citrullinemia type I
Answer: a
7. What is the primary symptom of urea cycle disorders?
a) Hyperammonemia
b) Hypoglycemia
c) Hypokalemia
d) Hyponatremia
Answer: a
8. Which amino acid is not produced by the urea cycle?
a) Arginine
b) Citrulline
c) Ornithine
d) Proline
Answer: d
9. Which of the following is not a cause of acquired hyperammonemia?
a) Liver failure
b) Drug toxicity
c) Excessive protein intake
d) Inherited urea cycle disorders
Answer: d
10. How is hyperammonemia treated in individuals with urea cycle disorders?
a) Restricting protein intake
b) Administering intravenous glucose
c) Taking ammonia-lowering medications
d) All of the above
Answer: d
11. Which molecule is required for the transport of ornithine into the mitochondria in the urea cycle?
a) ATP
b) ADP
c) GTP
d) GDP
Answer: a
12. What is the function of arginase in the urea cycle?
a) It converts argininosuccinate to arginine.
b) It converts arginine to urea.
c) It converts citrulline to argininosuccinate.
d) It converts ornithine to citrulline.
Answer: b
13. Which enzyme catalyzes the conversion of ammonia and bicarbonate to carbamoyl phosphate in the urea cycle?
a) Arginase
b) Carbamoyl phosphate synthetase I
c) Ornithine transcarbamylase
d) Argininosuccinate synthase
Answer: b
14. Which of the following is the primary nitrogen source for the urea cycle?
a) Glutamate
b) Aspartate
c) Glycine
d) Arginine
Answer: a
15. Which enzyme deficiency is associated with citrullinemia type I?
a) Arginase
b) Carbamoyl phosphate synthetase I
c) Argininosuccinate synthase
d) Argininosuccinate lyase
Answer: c
16. What is the primary role of the urea cycle in the body?
a) To convert ammonia to urea for excretion
b) To synthesize essential amino acids
c) To produce ATP for energy metabolism
d) To regulate blood glucose levels
Answer: a
17. Which of the following amino acids is not directly involved in the urea cycle?
a) Ornithine
b) Arginine
c) Citrulline
d) Aspartate
Answer: d
18. Which molecule acts as an allosteric activator of carbamoyl phosphate synthetase I?
a) N-acetylglutamate
b) Ornithine
c) Urea
d) Citrulline
Answer: a
19. Which enzyme catalyzes the conversion of argininosuccinate to arginine in the urea cycle?
a) Arginase
b) Carbamoyl phosphate synthetase I
c) Ornithine transcarbamylase
d) Argininosuccinate lyase
Answer: a
20. What is the fate of urea produced in the liver during the urea cycle?
a) It is excreted in urine.
b) It is converted to ammonia for energy production.
c) It is transported to the kidneys for filtration.
d) It is stored in the gallbladder for release during digestion.
Answer: a
21. Which of the following is an inherited disorder of the urea cycle?
a) Cirrhosis
b) Hepatitis
c) Uremia
d) Ornithine transcarbamylase deficiency
Answer: d
22. What is the primary consequence of a defect in the urea cycle?
a) Hyperammonemia
b) Hypoglycemia
c) Hyperglycemia
d) Hyperkalemia
Answer: a
23. In which organelle does the urea cycle occur?
a) Mitochondria
b) Nucleus
c) Endoplasmic reticulum
d) Golgi apparatus
Answer: a
24. Which of the following substances directly inhibits carbamoyl phosphate synthetase I?
a) N-acetyl glutamate
b) Arginine
c) Urea
d) Ornithine
Answer: c
25. Which of the following is a genetic disorder affecting the urea cycle?
a) Uremia
b) Hyperammonemia
c) Cystinuria
d) Phenylketonuria
Answer: b
26. Which enzyme deficiency is responsible for the most common urea cycle disorder?
a) Arginase
b) Carbamoyl phosphate synthetase I
c) Ornithine transcarbamylase
d) Argininosuccinate synthase
Answer: c
27. Urea cycle disorders primarily result in the accumulation of which toxic compound?
a) Urea
b) Ammonia
c) Glutamate
d) Aspartate
Answer: b
28. In individuals with urea cycle disorders, excessive ammonia levels primarily affect which organ?
a) Liver
b) Brain
c) Kidneys
d) Heart
Answer: b
29. Which of the following is not a symptom of hyperammonemia in urea cycle disorders?
a) Lethargy
b) Vomiting
c) Hypoglycemia
d) Seizures
Answer: c
30. How are urea cycle disorders diagnosed?
a) Genetic testing
b) Blood ammonia levels
c) Urine analysis
d) All of the above
Answer: d
31. Which of the following is a treatment option for urea cycle disorders?
a) Protein-rich diet
b) Liver transplantation
c) Ammonia-lowering medications
d) All of the above
Answer: b
32. What is the most severe form of urea cycle disorder?
a) Citrullinemia
b) Argininosuccinic aciduria
c) Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome
d) Ornithine transcarbamylase deficiency
Answer: d
33. Which of the following is an autosomal recessive urea cycle disorder?
a) Arginase deficiency
b) Citrullinemia type I
c) Argininosuccinate lyase deficiency
d) Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome
Answer: c
34. Which amino acid is essential for the synthesis of N-acetylglutamate, an activator of carbamoyl phosphate synthetase I?
a) Arginine
b) Ornithine
c) Lysine
d) Methionine
Answer: a
35. Which of the following is not a urea cycle intermediate?
a) Ornithine
b) Citrulline
c) Aspartate
d) Glycine
Answer: d
36. The enzyme deficiency in citrullinemia type I leads to the accumulation of which amino acid?
a) Arginine
b) Citrulline
c) Aspartate
d) Ornithine
Answer: a
37. Which urea cycle disorder is characterized by the accumulation of orotic acid?
a) Arginase deficiency
b) Citrullinemia type II
c) Ornithine transcarbamylase deficiency
d) Orotic aciduria
Answer: d
Assertion reasoning based question:
1. Assertion: Urea cycle disorders result in the accumulation of toxic ammonia in the body.
Reasoning: Urea cycle disorders are genetic conditions that impair the conversion of ammonia to urea, leading to hyperammonemia.
a) Both assertion and reasoning are correct, and the reasoning is a correct explanation of the assertion.
b) Both assertion and reasoning are correct, but the reasoning is not a correct explanation of the assertion.
c) Assertion is correct, but the reasoning is incorrect.
d) Assertion is incorrect, but the reasoning is correct.
Answer: a
2. Assertion: Urea cycle disorders primarily affect the liver.
Reasoning: The liver is the site where most of the urea cycle enzymes are produced and function.
a) Both assertion and reasoning are correct, and the reasoning is a correct explanation of the assertion.
b) Both assertion and reasoning are correct, but the reasoning is not a correct explanation of the assertion.
c) Assertion is correct, but the reasoning is incorrect.
d) Assertion is incorrect, but the reasoning is correct.
Answer: a
3. Assertion: Hyperammonemia can lead to neurological symptoms in urea cycle disorders.
Reasoning: Excessive ammonia levels can cause damage to the central nervous system, resulting in cognitive impairment and seizures.
a) Both assertion and reasoning are correct, and the reasoning is a correct explanation of the assertion.
b) Both assertion and reasoning are correct, but the reasoning is not a correct explanation of the assertion.
c) Assertion is correct, but the reasoning is incorrect.
d) Assertion is incorrect, but the reasoning is correct.
Answer: a
4. Assertion: Restricting protein intake is a common dietary intervention for managing urea cycle disorders.
Reasoning: Dietary protein contains nitrogen, which can lead to increased ammonia production in individuals with urea cycle disorders.
a) Both assertion and reasoning are correct, and the reasoning is a correct explanation of the assertion.
b) Both assertion and reasoning are correct, but the reasoning is not a correct explanation of the assertion.
c) Assertion is correct, but the reasoning is incorrect.
d) Assertion is incorrect, but the reasoning is correct.
Answer: a
5. Assertion: Liver transplantation can be a potential treatment option for severe urea cycle disorders.
Reasoning: Liver transplantation provides a source of functional urea cycle enzymes, which can restore the normal metabolism of ammonia.
a) Both assertion and reasoning are correct, and the reasoning is a correct explanation of the assertion.
b) Both assertion and reasoning are correct, but the reasoning is not a correct explanation of the assertion.
c) Assertion is correct, but the reasoning is incorrect.
d) Assertion is incorrect, but the reasoning is correct.
Answer: a
6. Assertion: Urea cycle disorders are exclusively inherited and cannot be acquired.
Reasoning: Urea cycle disorders result from genetic mutations that affect the enzymes involved in the urea cycle.
a) Both assertion and reasoning are correct, and the reasoning is a correct explanation of the assertion.
b) Both assertion and reasoning are correct, but the reasoning is not a correct explanation of the assertion.
c) Assertion is correct, but the reasoning is incorrect.
d) Assertion is incorrect, but the reasoning is correct.
Answer: a